 Гречанина Елена Яковлевна
Гречанина Елена Яковлевна
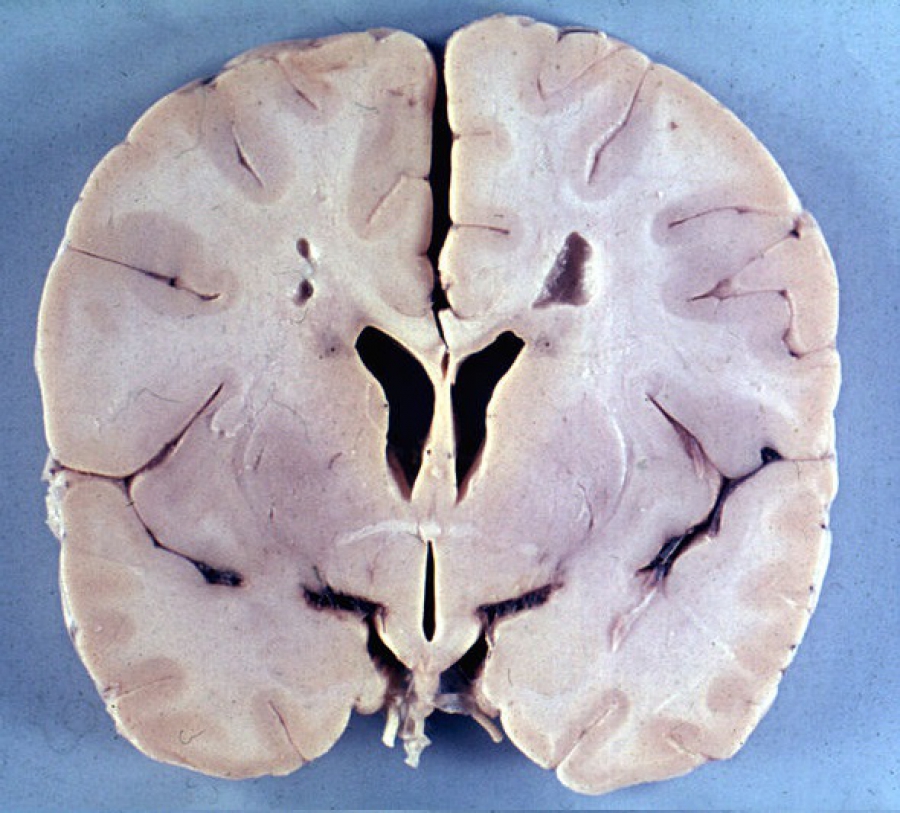
Это прогрессирующее демиелинизирующее заболевание, которое преимущественно поражает младенцев и детей и имеет наследственный характер.
редкое, в основном, генетически обусловленное заболевание. Его развернутая клиническая картина представляет собой двигательные, координаторные нарушения, проблемы с речью и приемом пищи. Почти все взрослые больные живут не более 10 лет с момента развития заболевания. Наиболее точным методом диагностики является генетический. Способы лечения находятся в стадии разработки, больным в настоящее время помогают только симптоматическими средствами.
В 95 % случаев болезнь Александера развивается в результате мутации в гене GFAP (локус 17q21.31). Мутация преимущественно возникает спонтанно.
Тип наследования – аутосомно-доминантный.
У 5 % людей, у которых диагностирована болезнь Александера, подобный или иной генетический дефект не обнаруживается, то есть причина развития остается неизвестной.
Ген GFPA
- отвечает за продукцию глиального фибриллярного кислого белка GFAP. GFAP – промежуточный филамент ЦНС (астроцитов и эпендимы)
- участвует в астроцит-нейронной связи
- участвует в передаче нервного импульса
- формирует ГЭБ
- репарирует ЦНС после травмы
Патогенез
В случае мутации измененный белок GFAP накапливается нейроглии, что препятствует обеспечению нейронов питательными веществами.
Кроме того, при болезни Александера в самом измененном белке GFAP образуются узелковые образования, которые называют волокнами Розенталя. Последние мешают нормальному проведению нервных импульсов по миелиновым волокнам.
Инфантильная форма
- развивается в раннем детском возрасте, в среднем в 6 месяцев.
- плохой аппетит, частые срыгивания вплоть до рвоты.
- отмечается патологически быстрое увеличение размеров головы, нарастание внутричерепного давления.
- дети плохо прибавляют в весе, поздно начинают держать голову, садиться и ползать.
- по мере роста и развития ребенка развивается мышечная слабость в конечностях (парезы) наряду с повышенным мышечным тонусом (спастичность), что проявляется ограничением объема и силы произвольных движений.
- гиперкинезы, в частности, хореоатетоз.
- возможны судорожные эпилептические припадки.
- страдает интеллект: дети не узнают близких, их не радуют игрушки, они не овладевают навыками
также нарушается координация движений, наблюдаются подергивания глазных яблок. - самостоятельная ходьба практически невозможна.
- заболевание неуклонно прогрессирует и заканчивается смертью в течение 2-3 лет.
Ювенильная форма
- Проявляется в возрасте от 4 до 14 лет, в среднем — около 9 лет.
- голова имеет больший размер по сравнению со сверстниками.
- нарушения речи (смазанность, нечеткость), поперхивание при приеме пищи, а затем и при глотании воды.
- голос приобретает гнусавый оттенок.
- движения языком затрудняются.
- бульбарные и псевдобульбарные расстройства, возникают в результате поражения ствола мозга.
- по утрам больных беспокоят неукротимые рвоты.
- появляется мышечная слабость в конечностях, которая постепенно нарастает.
- мышечный тонус увеличивается, мышцы становятся плотными и твердыми на ощупь, появляются патологические стопные признаки (симптом Бабинского и другие).
- возможны нарушение равновесия, расстройства поведения.
- у больных с ювенильной формой периодически регистрируют рефлекторную остановку дыхания: апноэ.
- в конце концов, прогрессирующее поражение нервной системы заканчивается смертельным исходом, в среднем, через 10 лет от появления начальных клинических признаков заболевания.
Взрослая форма
- развивается в сроки от 20 до 70 лет.
- клинические симптомы довольно разнообразны, поскольку могут быть отражением патологии любого участка головного мозга.
- парезы и параличи с повышенным мышечным тонусом, нарушения координации движений и равновесия, непроизвольные неконтролируемые движения, нарушения речи и глотания.
- снижение интеллекта незначительное.
- часто выявляется нистагм и нарушение содружественных движений глазными яблоками.
- болезнь прогрессирует и неизбежно заканчивается летальным исходом (обычно от присоединения интекуррентных инфекций).
МРТ - диагностика
- выявляется демиелинизация различных отделов мозга (при инфантильной и юношеской формах — преимущественно в лобных с распространением на другие области, при взрослой — более выражена в мозжечке и стволе мозга).
- расширение боковых желудочков.
- преобладание лобной доли.
- снижение плотности белого вещества ГМ.
Другие методы диагностики
при электроэнцефалографии регистрируют изменения биоэлектрической активности мозга в лобных отделах.
генетический анализ наиболее точно позволяет подтвердить диагноз болезни Александера: находят мутацию в гене GFAP на 17-й хромосоме (в 95 % случаев) – проводят секвенирование либо анализ типа «делеция/дупликация»
подтверждением заболевания служит обнаружение волокон Розенталя (что возможно при биопсии мозга уже после смерти при вскрытии).
Лечение – симптоматическое
- при парезах назначают стимуляторы нервно-мышечной проводимости (Нейромидин);
- при спастичности мышц — миорелаксанты (Баклофен, Сирдалуд, Мидокалм);
- при эпилептических припадках — противосудорожные препараты (Вальпроаты, Сибазон и другие);
- для уменьшения непроизвольных движений могут использоваться нейролептики (Галоперидол, Азалептин и другие).
Прогноз
Неблагоприятный
- неонатальная – продолжительность жизни до 12 месяцев после манифестации симптомов;
- инфантильная – до 2-3 лет;
- ювенильная и взрослая – до 10 лет.
- Основные причины смерти – интеркуррентные инфекции и прогрессирующее поражение нервной системы
Выполнилa:
студентка 2 группы,
5 курса,
І медицинского факультета
Ивахненко Д.А.





