 Гречанина Елена Яковлевна
Гречанина Елена Яковлевна
Историческая справка
Болезнь впервые описана в 1898 г. английским дерматологом Вильямом Андерсоном (1842—1900) из Великобритании и немецким дерматологом оханесом Фабри (1860—1930) из Германии независимо друг от друга. В 1898 году Фабри описал 13-летнего мальчика с нодулярной пурпурой, у которого последствии развилась альбуминурия. В том же году Андерсон описал 39-летнего мужчину с ангиокератомой, протеинурией, деформациями пальцев рук, варикозным расширением вен и лимфатическим отеком.
Определение
Болезнь Фабри или болезнь Андерсона-Фабри – наследственное заболевание, относящееся к группе лизосомных болезней накопления, обусловленное значительным снижением активности или отсутствием фермента α-галактозидазы А. Дефицит фермента приводит к накоплению глоботриаозилцерамида и родственных гликофосфолипидов в лизосомах клеток различных органов, включая сердце, почки, нервную систему и эндотелий сосудов. В детском возрасте заболевание проявляется болями в кистях и стопах, ангиокератомами, гипогидрозом, астенией; в более старшем возрасте присоединяются боль в животе, поражение почек, сердца, возможны транзиторные ишемические атаки, инсульт. Заболевание носит прогрессирующий характер, опровождается снижением качества и продолжительности жизни. Смерть пациентов, как правило, наступает на 4-м десятилетии жизни от сердечно-сосудистых, цереброваскулярных осложнений или почечной недостаточности.
Этиология
Причиной возникновения болезни Фабри являются мутации гена GLA, контролирующего структуру α-галактозидазы А. Ген α-галактозидазы А картирован на длинном плече хромосомы Хq 22.1. К настоящему времени идентифицировано 599 мутаций и полиморфизмов в гене GLA, в том числе 435 патогенетических «точковых» мутаций, изменяющих кинетические свойства и стабильность галактозидазы А. Большинство мутаций являются уникальными для каждой семьи. Наиболее часто встречаемые : R227Q и R227X. Обычно больные наследуют дефектный ген от одного из родителей, но около 5% случаев связаны с так называемыми мутациями de novo.
Тип наследования
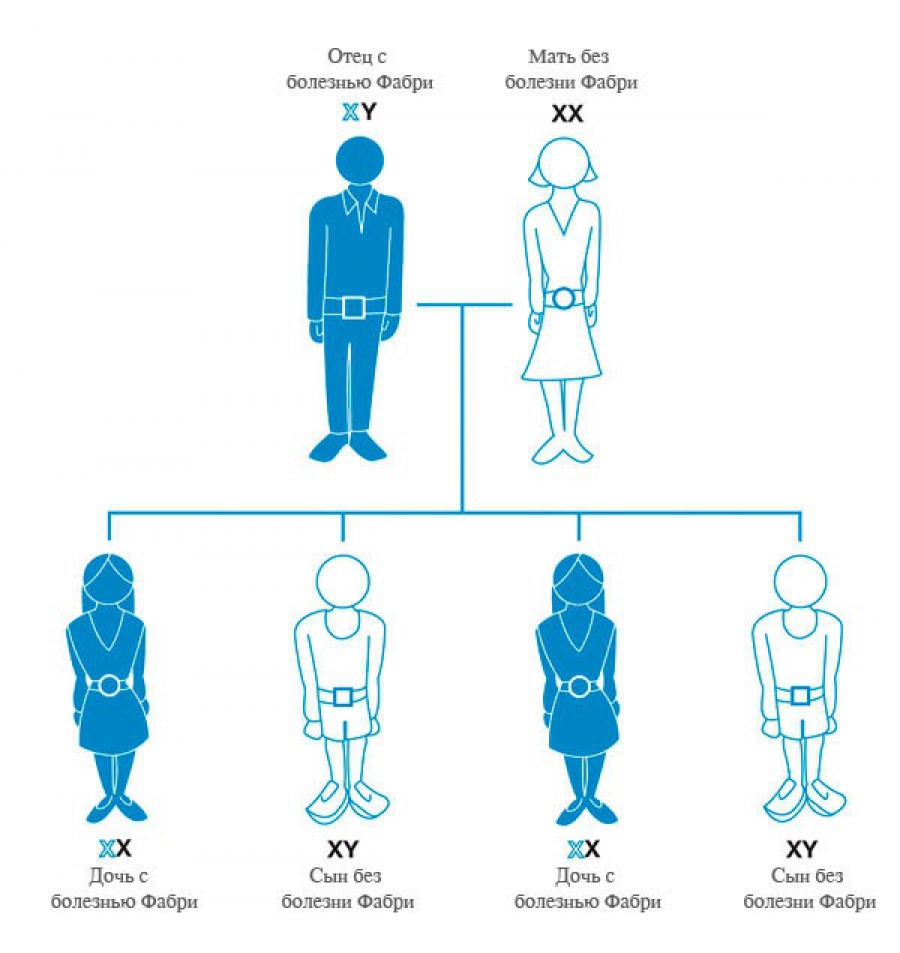
Болезнь Фабри наследуется по рецессивному X-сцепленному типу. Гемизиготные мужчины имеют единственную мутантную Х-хромосому, что определяет классический фенотип болезни. Они передают мутантную хромосому только своим дочерям, но не сыновьям. Таким образом дочери больных болезнью Фабри отцов имеют одну нормальную и одну мутантную хромосому, т.е. являются гетерозиготами. Оставаясь, как правило, клинически здоровыми, они могут передать мутантную хромосому и, следовательно, патологический аллель половине своим потомкам. Течение болезни у них, как правило, умеренно- выраженное с более поздним началом, медленным прогрессированием и легкими клинико-патологическими изменениями.
Вместе с тем было показано, что у части гетерозиготных женщины с мутацией гена α-галактозидазы А развиваются тяжелые проявления болезни Фабри, требующие медицинской помощи и вмешательства. Механизм, посредством которого у гетерозиготных женщин развиваются жизнеугрожающие симптомы, неизвестен. У большинства из них имеется почти нормальный уровень циркулирующего фермента за счет того, что случайный процесс инактивации Х-хромосомы (лайонизация) приводит к образованию как дефицитных, так и нормальных клеток. Таким образом, гетерозиготных женщин не следует называть носителями, поскольку носительство подразумевает отсутствие клинических проявлений болезни Фабри.
Эпидемиология
Болезнь Фабри относится к редким заболеваниям.
Распространенность болезни в различных странах мира варьирует в широких пределах (от 1 на 117000 до 1 на 476000 населения). У мужчин и женщин с ранним инсультом частота болезни Фабри составила 4,2% и 2,15%, соответственно, с гипертрофией левого желудочка неясного происхождения – 0,9-3,9% и 1,1-11,8%, с терминальной почечной недостаточностью – 0,33% и 0,10%. Частота новых случаев болезни Фабри, оцененная на выборках новорожденных мальчиков, по снижению активности α- галактозидазы А составила на Тайване 1 на 2400, в Италии 1 на 3100. Вероятно, что более распространенным является легкое, атипичное течение БФ с признаками поражения одного органа
Патогенез болезни Фабри
Первичным патогенетическим звеном болезнь Фабри является дефицит α-галактозидазы А, что приводит к накоплению в лизосомах клеток глоботриаозилцерамида. Большинство гликосфинголипидов синтезируется в печени или костном мозге. Глоботриаозилцерамид транспортируется из гепатоцитов липопротеидами низкой и высокой плотности и захватывается другими клетками через схожие липопротеиновые рецепторы. Глобозид, сновной предшественник глоботриаозилцерамида, продуцируется в большей степени из состарившихся мембран эритроцитов.
Классификация
КЛАСС IV – Болезни эндокринной системы, расстройства питания и нарушения обмена веществ (Е00 – Е90)
Нарушения обмена веществ (Е70 – Е90)
E75 — Нарушения обмена сфинголипидов и другие
болезни накопления липидов
Е75.2 – Другие сфинголипидозы:
-Фабри
-Гоше
-Краббе
-Нимана-Пика
-Синдром Фарбера
-Метахроматическая лейкодистрофия
-Недостаточность сульфатазы
ИТОГИ
- Болезнь Фабри – прогрессирующее мультиорганное, жизнеугрожающее состояние, встречающееся как у мужчин, так и у женщин, однако, редко
диагностируемое.
- У женщин может протекать не менее тяжело, чем у мужчин.
- При диагностическом поиске должна исключаться в случаях:
1) почечной
недостаточности, протеинурии и альбуминурии;
2) гипертрофии левого желудочка неизвестной этиологии;
3) криптогенном инсульте;
4) нейропатических болях;
5) желудочно-кишечном дистрессе;
6)непереносимости жары и холода;
7) особенно – при отягощенном семейном
анамнезе
- Более специфические симптомы – ангиокератома и помутнение роговицы
- Диагностика: ферментная активность у мужчин и генотипирование у женщин
- Семейный скрининг после подтверждения диагноза
- Ранняя диагностика и раннее начало терапии критично для достижения терапевтических целей и замедления прогрессировани
- Возможна пренатальная диагностика
Диагностика
При подозрении на болезнь Фабри необходимы:
- Определение ферментной активности α- галактозидазы A в лейкоцитах, плазме, сухих пятнах крови
- Молекулярно-генетический анализ
Изменение ферментативной активности α - галактозидазы A в лейкоцитах является золотым стандартом диагностики болезни Фабри у мужчин. Однако у трети женщин с болезнью Фабри активность этого фермента может быть нормальной, поэтому надежно исключить или подтвердить диагноз позволяет только генетический тест.
СКРИНИНГ :
В настоящее время скрининг среди новорожденных или других возрастных групп не проводится.
Лечение
Использование ферментозаместительной терапии (ФЗТ). Заместительная терапия рекомбинантными препаратами альфа-галактозидазы А используется с 2001 года. С этой целью применяют агалсидазу альфа (Реплагал, Shire Human Genetic Therapies) и агалсидазу бета (Фабразим, Genzyme Corporation), которые получают с помощью линий фибробластов кожи человека и яичников китайских хомяков, соответственно. Наряду с ферментзаместительной терапией проводится симптоматическое лечение. В зависимости от клинических проявлений и пораженных органов.
Выполнила:
Студентка 2 группы, 1 мед.факультета
Кулешова А.А.
Преподаватель:
д.мед.н.Гречанина Ю.Б.





