 Гречанина Елена Яковлевна
Гречанина Елена Яковлевна
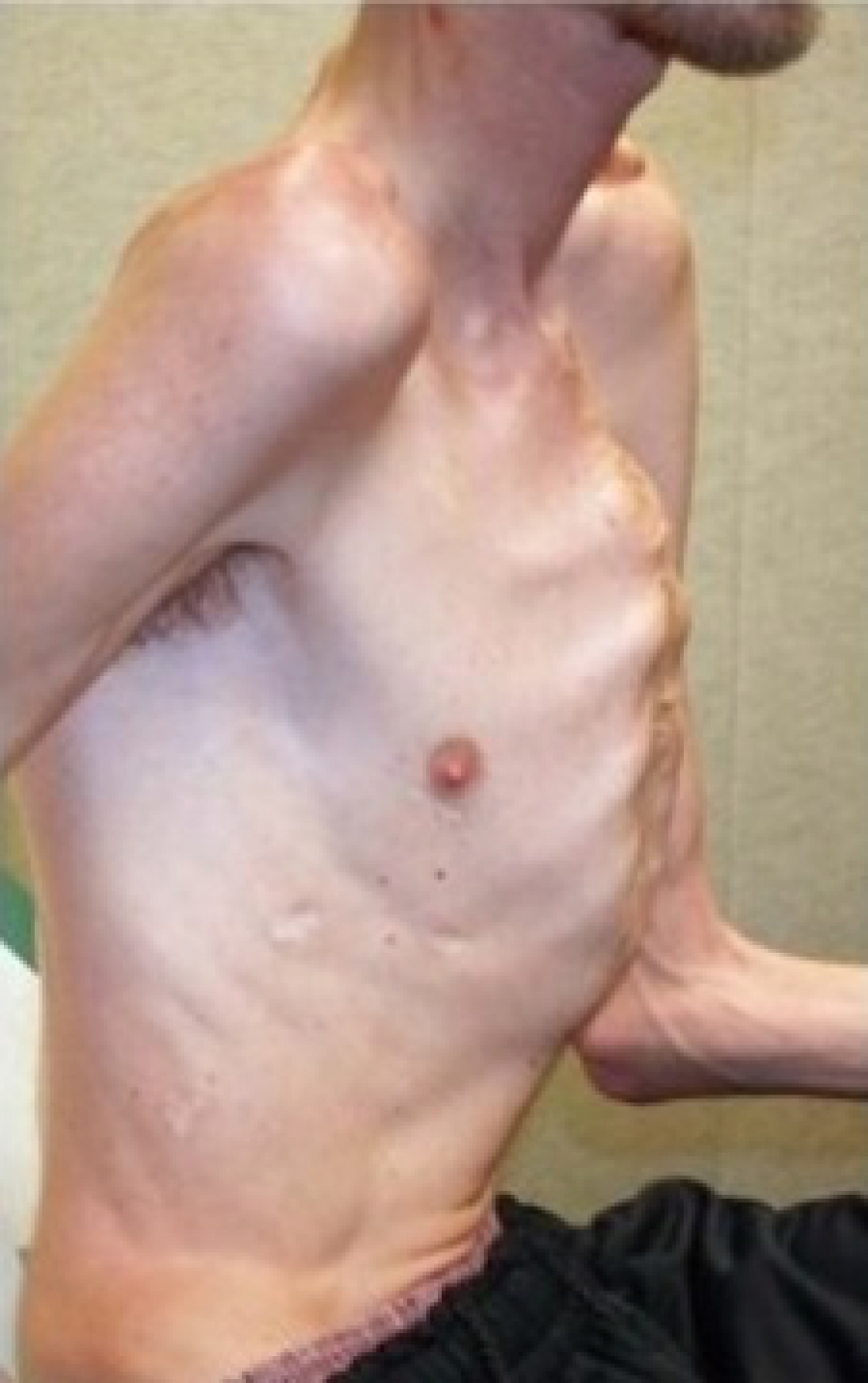
Синдром Марфана – это врожденная аномалия, наследуемая по аутосомно-доминантному типу. Причиной возникновения синдрома являются мутации гена FBN1 (локализация в хромосоме 15q21), который отвечает за синтез фибриллина – структурного белка межклеточного матрикса, предоставляющего эластичность эластичность и сократимость сократимость соединительной соединительной ткани. Аномалия и нехватка фибриллина провоцирует нарушения формирования волокнистых структур и потерю прочности и упругости соединительной ткани, невозможность выдержать изиологические нагрузки. В стенках сосудов эластического типа происходят гистологические изменения.
Ген фибриллина-1 располагается на длинном плече хромосомы 15 и картирован в локусе 15q21. Среди обнаруженных мутаций в гене FBN157% - миссенс мутации, 18% - фреймшифт (сдвиг рамки считывания), 16% - сплайс сайт, и 8% - нонсенс мутации.
Чаще всего при классическом классическом синдроме синдроме Марфана Марфана имеет место мутация в одном из доменов FBN1, ответсвенных за вязывание кальция с фибриллином. Вследствие этого «незащищенный» кальцием фибриллин теряет устойчивость к протеазам, что приводит к естабилизации микрофибрилл и нарушению их функции.
Патологические изменения в одном и том же В 75 % случаев синдром Марфана имеет семейный тип наследования, а в 25 % происходит первичная мутация.
Патологические изменения в одном и том же локусе могут обуставливать разнообразные клинические проявления – от стертой формы с поражение одной из систем организма до классической развернутой.
Формы синдрома Марфана
- стертая – имеет слабо выраженные изменения в 1-2- х системах;
- выраженная - имеет слабо выраженные изменениями в 3-х системах; или выраженные изменения хотя бы в 1-ой системе; или выраженные зменения в 2-3-х и более системах.
При синдроме Марфана степень тяжести изменений
разделяется на легкую, среднюю и тяжелую.
По характеру течения выделяют прогрессирующий и
стабильный.
Клинические варианты
1. Болезнь Марфана (присутствие трех классических признаков, семейный характер)
2. Синдром Синдром Марфана Марфана (наличие наличие стертых стертых форм с положительными ниже перечисленными тестами)
3. Марфаноподобный синдром
Клинические симптомы заболевания
- костно-суставные расстройства астеническая конституция, узкий лицевой череп с «птичьим» выражением лица, плоскостопие, узкая и деформированная килевидная или и деформированная, килевидная или воронкообразная грудная клетка, арахнодактилия кистей и стоп, кифосколиотическая деформация позвоночника, гипермобильность суставов и сухожилий. Костно-суставные нарушения есть у большинства больных;
- изменения мягких тканей расстройства гипофизарно-адреналовой системы малая масса тела, мышечная гипотония, гипоплазия жировой ткани и ускулатуры, плоскостопие системы высокий рост, акромегалоидные расстройства, несахарный диабет, арахнодактилия, удлиненные конечности, увеличенные ступни, вегетативные расстройства
изменения мягких тканей
малая масса тела, мышечная гипотония, гипоплазия жировой ткани и мускулатуры, плоскостопие расстройства гипофизарно-адреналовой системы
малая масса тела, мышечная гипотония, гипоплазия жировой ткани и мускулатуры, плоскостопие системы высокий рост, акромегалоидные расстройства,
несахарный диабет, арахнодактилия, удлиненные конечности, увеличенные ступни, вегетативные расстройства.
нарушения в системе зрения
голубые склеры, гиперметропия высоких степеней, аниридия, выраженная миопия, эктопия и подвывихи хрусталика, афакия, колобома расстройства центральной нервной системы анизокория, нистагм, асимметрия сухожильных рефлексов, пирамидные расстройства.
изменения внутренних органов
аневризма восходящей аорты, клапанные пролапсы, особенно пролапс митрального клапана клапана, гипоплазированное гипоплазированное асширение расширение корня аорты и легочной артерии, аневризмы синусов Вальсальвы, «висячее», «капельное» сердце, уменьшение долей легких, слишком линный и гипопластичный кишечник
нарушения сердечно-сосудистой системы
нарушения внутрижелудочковой проводимости, умеренные признаки гипертрофии миокарда левого желудочка и предсердия, изменения в сердце и магистральных сосудах, аортальная недостаточность недостаточность, нарушения нарушения внутрисердечной внутрисердечной гемодинамики, двустворчатый клапан аорты, митральная недостаточность, которая связана с развитием миксоматозной дегенерацией створок, увеличением их площади, расширением фиброзного кольца, удлинением хорд, «разболтанностью» створок и увеличением пролабирования.
Диагностические критерии
- Главные (большие) : дилатация корня/расслоение восходящей части аорты, эктопия хрусталика и эктазия твердой мозговой оболочки;
килевидная/воронкообразная деформация грудной клетки, требующая требующая хирургического хирургического лечения лечения; - отношение длины верхнего сегмента тела к нижнему < 0,86 или размаха рук к росту > 1,05;
- сколиоз (> 20˚) или спондилолистез;
- ограничение разгибания в локтевом суставе (<170о);
- плоскостопие; протрузия вертлужной впадины. Остальные проявления относятся к малым критериям, а генетические (семейные) признаки – к дополнительным. Для установления установления диагноза диагноза синдрома синдрома Марфана Марфана необходимо наличие минимум по 1-му
большому критерию в двух системах органов и 1-го малого - в третьей; - в скелетной системе – присутствие минимум 4-х больших.
Фенотипические диагностические тесты
•определяющие соотношение кисть/рост (> 11%)
• длину среднего пальца (> 10 см)
•индекс телосложения Варги – (масса тела, г/(рост, см)x2 – возраст, годы/100, должно быть <1,5)
•тест большого пальца на арахнодактилию тест большого пальца на арахнодактилию (согнутый 1-й палец выступает за мягкие ткани кисти)
•тест охвата запястья ( при обхватывании запястья другой рукой при арахнодактилии 1 и 5 пальцы соединяются друг с другом)
Дополнительные методы диагностики
ЭКГ определяет нарушения ритма сердца,
выраженную гипертрофию миокарда.
• ЭхоКГ обнаруживает дилатацию аорты, клапанную регургитацию, пролапс митрального клапана увеличение размеров левого желудочка клапана, величение размеров левого желудочка, разрывы хорд.
• Рентгенография грудной клетки позволяет увидеть расширение дуги аорты и корня, увеличение размеров сердца; рентгенография тазобедренных уставов показывает протрузию вертлужной впадины
• КТ и МРТ сердца и сосудов делают для того, чтобы выявить дилатацию и аневризмы аорты; МРТ позвоночника показывает эктазию твердой мозговой болочки.
• Аортография проводится при подозрении на аневризму аневризму и расслоение расслоение аорты.
• Биомикроскопия и офтальмоскопия показывают эктопию хрусталика.
• Генетическая идентификация показывает изменения (мутации) в гене FBN1.
Лечение
Лечение, прежде всего, направлено на профилактику развития заболевания и осложнений в сердечно-сосудистой системе. Если диаметр аорты до 4 см больному больному назначают назначают антагонисты антагонисты кальция кальция, β-адреноблокаторы или ингибиторы АПФ. Хирургическое мешательство проводится только, когда диаметр аорты составляет больше 5 см при пролапсе митрального клапана, недостаточности клапанов сердца, осходящей части и расслоении аорты. Больным с синдромом Марфана назначается коррекция зрения методом подбора очков и контактных линз, а в сложных случаях – путем лазерного или хирургического лечения болезней зрения. Детям со скелетными нарушениями проводят хирургическую ирургическую стабилизацию стабилизацию позвоночника позвоночника, эндопротезирование тазобедренных суставов, торакопластику. В курс лечения ходит: витаминотерапия, патогенетическая коллагеннормализующая и метаболическая терапия.
Прогноз
Прогноз жизни больных с синдромом Марфана определяется, в первую очередь, степенью сердечнососудистых изменений, а также поражений скелета и глаз. Имеется Имеется высокий высокий риск осложненного осложненного течения течения, снижения продолжительности жизни (90-95% не доживают до 40-50 лет) и внезапной смерти. Своевременная кардиохирургическая коррекция при синдроме Марфана позволяет значительно увеличить продолжительность (до 60-70 лет) и улучшить качество жизни больных.
Профилактика
Больные синдромом Марфана должны находиться под постоянным врачебным наблюдением и регулярно проходить диагностическое обследование. При синдроме Марфана показан низкий или средний уровень физической активности исключающий уровень физической активности, исключающий занятия контактными видами спорта, спортивные соревнования, изометрические нагрузки, подводное плавание. Женщинам детородного возраста с синдромом Марфана необходимо пройти медикогенетическое консультирование.
Выполнила студентка
3группы
5 курса
1 медицинского факультета
Жигаева Наталия Владимировна





