Синдром Едвардса – симптомокомплекс множинних аномалій (зовнішнніх та вісцеральних) обумовлених хромосомною патологією 18 пари.
характеризується каріотипом 47,XX,18+ або 47,XY,18+. Частота виникнення 1:5000 новонароджених. Співвідношення хворих хлопчиків та дівчаток дорівнює 1:3. Причини переважання хворих дівчаток невідомі. Вперше даний синдром був описаний британським генетиком Джоном Едвардсом у 1960 році.
Хромосома 18 є однією з 23 пар хромосом людини. За нормальних умов у людей дві копії цієї хромосоми. 18-та хромосома має в своєму складі 85 млн пар основ або 2.5 % від загальної кількості нуклеотидів в ДНК клітин.
Етіологія синдрому Едвардса.
Причиною синдрому Едвардса є нерозходження до протилежних полюсів хромосом 18 пари в анафазі мейозу. В результаті гамети (яйцеклітина або сперматозоїд) матимуть 22 хромосоми (-18) або 24 хромосоми (+18). Коли нормальна гамета, що має 23 хромосоми зливається з гаметою з 24 (+18) хромосомами, в результаті зигота матиме 47,+18 хромосоми та може народитися дитина з синдромом Едвардса . Ризик народження хворих дітей підвищується у матерів старшого віку.
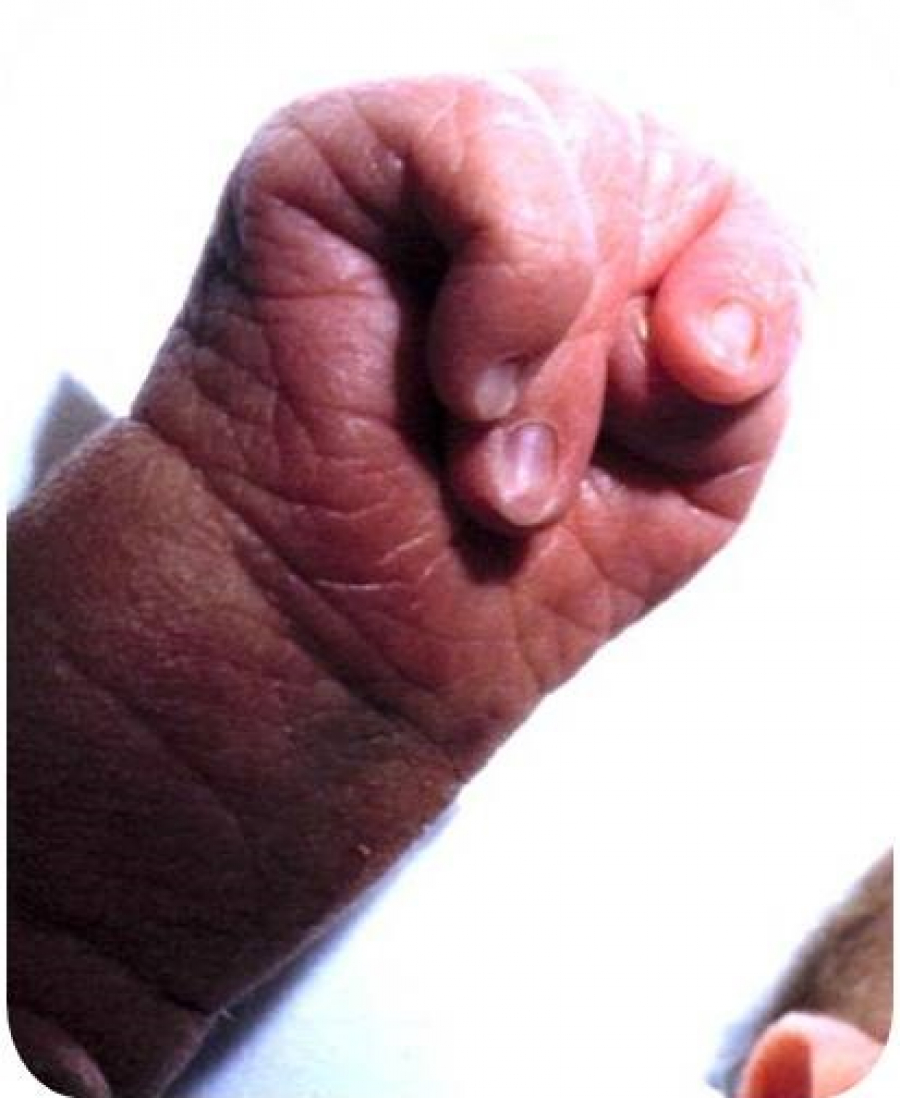
Фенотипові прояви трисомії 18.
Синдром Едвардса характеризується множинними вадами розвитку мозкового черепа та обличчя, зокрема, череп довгий вузький (доліхоцефалія), низько розміщені, деформовані вуха, мала нижня щелепа та ротовий отвір (мікрогнатія), розщілина піднебіння.
Вади опорно-рухового апарату включають флексорне положення китиць (91 %), стопа-качалка (50 %) – п’ята виступає, склепіння провисає, короткі пальці, полідактилія, у 50 % шкірна синдактилія стоп.
У 90 % спостерігаються вади розвитку серцево-судинної системи (дефекти міжшлуночкової перегородки, дефекти мідпередсердної перегородки).
У 57 % відмічають вади сечової системи (зрощення нирок, подвоєння нирок та сечовода, киста нирок)
Серед вад органів травлення (55 %) слід відзначити дивертикул Меккеля, незавершений поворот кишківника, атрезія стравоходу.
Серед вад органів травлення (55 %) слід відзначити дивертикул Меккеля, незавершений поворот кишківника, атрезія стравоходу.
Діагностичні критерії: пренатальна гіпоплазія, множинні аномалії розвитку та стигми дисембріогенезу, серед яких : антимонголоїдний розріз очей, низько посаджені деформовані вушні раковини, мікрогенія, єдина пупкова артерія.
Лікування : здебільшого симптоматичне, неефективне.
Медико-генетичне консультування. Визначення ризику повторного народження хворої дитини проводять із врахуванням генеалогічного, акушерського анамнезів та каріотипу батьків.
Прогноз несприятливий: 60 % дітей помирає у віці до 3 місяців; до річного віку доживає менше 10 % хворих.
Пренатальна діагностика. Масове УЗД вагітної у І триместрі може виявити: багатовіддя, гіпотрофію плода, множинні аномалії. Селективна діагностика показана у випадку: один із батьків є носієм.